
|
新方法能更精确计算大分子间范德华力时间:2025-10-31 奥地利维也纳理工大学科学家对以往计算分子间作用力的方法进行了改进,新方法在计算大分子之间的范德华力时更加精确。相关研究成果发表于新一期《自然·通讯》。
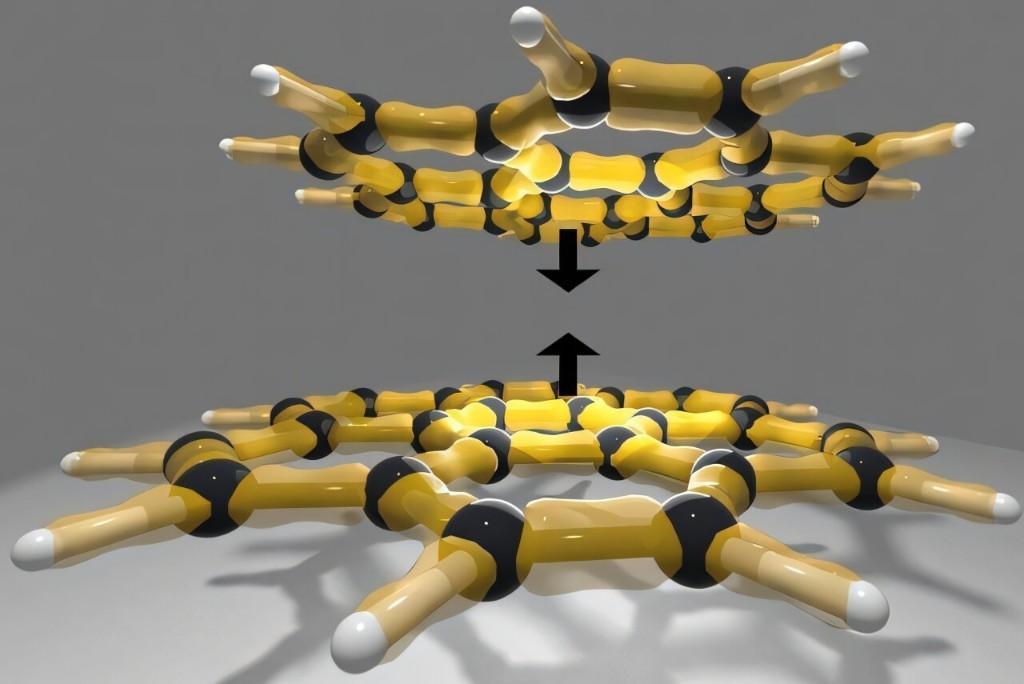
从壁虎爬墙,到氮气在-196℃时液化,许多日常现象都离不开范德华力的作用。范德华力的精确计算在材料科学、药物研发等领域至关重要。无论是理解药物在片剂中的结晶过程,还是评估材料储存氢能的能力,都离不开对分子间作用的准确模拟。 然而,这种分子间微弱的作用力一直难以精确计算,不同方法得出的结果常常相互矛盾,成为长期困扰学界的难题。 研究团队对目前最常用的两种方法进行了比较:一种是量子蒙特卡罗模拟,通过计算机模拟无数可能的电子排布;另一种是耦合簇方法,先处理分子的低能状态,再引入高能构型作为修正。 耦合簇方法一直被视为“金标准”,但团队发现,与蒙特卡罗模拟相比,该方法存在微小却持续的系统性偏差,原因多年来未能找到。 如今,团队找到了答案:耦合簇方法会系统性地高估大分子和高极化分子的结合能。他们对此方法进行了改进,在不显著增加计算成本的前提下纠正了偏差,使结果与量子蒙特卡罗数据更为吻合。 新方法大幅降低了大分子系统的计算负担。过去,对含有一百个以上原子的分子进行模拟,计算量极为庞大,如今这一难题迎刃而解。 研究团队表示,这一进展不仅有助于科学家更准确地预测大分子行为,也为深入理解生物系统、推动可再生能源技术发展奠定了基础。此外,新方法所提供的高可靠性数据,还可作为人工智能模型的训练样本,用于虚拟环境中新材料与药物的设计。 |
山西科普网是一个集原创数字化作品传播、科普活动展示、科普资源库下载、互动交流于一体的科普网站,秉承严谨求实的科学态度,
打造全国一流的科普信息传播平台。
合作机构
山西省科学技术协会 山西科技新闻出版传媒集团 深圳科普网 山西科技展教中心
联系我们
电话/TEL
0351—7041988
地址/ADDRESS
山西省太原市万柏林区晋祠路23号

